This page describes
tools and guidelines for use in assigning newly identified Hops
to existing families and subgroups. These protocols are written
for researchers whose expertise in phylogenetic analyses does
not extend beyond basic BLAST analyses. The general approach involves
alignment of sequences in a given Hop family using ClustalW followed
by use of MEGA2.1 to evaluate clustering and calculate genetic
distance.
NOTE
2-12-10: Since publication of Lindeberg et al., 2005, MEGA 2.1 has been replaced with MEGA 4. The following tutorial uses MEGA 4.
Note that
guidelines for selecting the Hop names themselves including names
for novel Hops, Hops belonging to novel or preexisting subgroups
in previously identified families, as well as chimeric, truncated,
or non-expressed Hops can be found on the Name
structure and Selection page.
Outline
of procedures for Hop phylogenetic analysis:
(download
pdf of this protocol)
I.
Confirm similarity to one or more previously characterized Hops
using BLAST analysis
II. Sequence alignment in ClustalW
A.
Obtain
a file listing all sequences in the Hop family of interest
B. Generate alignment file
III.
Clustering analysis and genetic distance calculation in MEGA
A.
Download and install MEGA
B. Convert Clustal alignment file to
MEGA format
C. Perform clustering analysis in MEGA
D. Calculate genetic distance between
the new Hop and established subgroups
I.
Confirm similarity to one or more previously characterized Hops
using BLAST analysis.
- Conduct
BLASTP analyses to determine whether a given protein is similar
to any previously characterized Hops.
- If
there IS NO significant similarity to one or more previously
characterized Hops, and and the newly identified Hop has been
confirmed by criteria other than sequence similarity (see
Criteria for Hop name assignment),
go to Name structure and Selection:
How to name a Hop for guidelines on naming novel Hop proteins.
- If
there IS significant similarity to one or more previously
characterized Hops (roughly defined as a BLAST expect value
of less than 10-5 and with alignment
extending over 60% of the length of the protein) follow the
steps below to assign a subgroup classification, or contact
the PPI site administrator
and a subgroup classification can be generated for you.
II.
Sequence alignment in ClustalW
A.
Obtain a file listing all sequences in the Hop family of interest
- Go to
the list of assigned Hop names
and obtain the sequences for all members of the appropriate
family by clicking on the family name.
- Save
the list of sequences as a text file. Note that the sequences
are listed in FASTA
format.
- Add the
sequence of the newly identified Hop to the list (also in
FASTA format) and save the file.
B.
Generate alignment file
ClustalW
is a general purpose tool for alignment of multiple sequences.
It is available through numerous websites, including the EMBL-EBI
site described here. (The HopA family is used to illustrate
the various procedures)
- Go to
ClustalW
at EMBL-EBI (now Clustal Omega):
- Paste or upload your protein sequences in fasta format into the specified window. Parameters can stay at their default settings. Click
on the link to download the alignment file. Save as a text file, renaming
it if desired
III.
Clustering analysis and genetic distance calculation in MEGA.
MEGA (Molecular Evolutionary Genetics Analysis) is a free software
package for comparative sequence analysis
A.
Download and install MEGA
- Go to
the MEGA
4 download site, provide the requested information,
and download the program. An .exe file will appear on your
desktop.
- Open
the .exe file and follow the instructions for installation
of MEGA 4.
B.
Convert Clustal alignment file to MEGA format
- Open
the installed MEGA program. The following window will appear
- Under
File on the menu select "Convert to MEGA format".
The following window will appear:
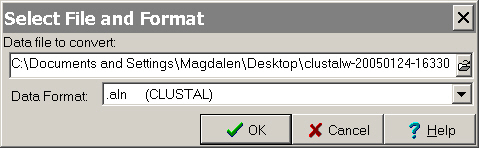
- Select
the output file you saved from ClustalW as "Data file to convert:"
and for "Data format" select ".aln (CLUSTAL)".
Click OK.
The converted file will appear in the "Text File Editor
and Format Converter" window.
- Scroll
through the converted file to check format.
If line numbers are present, either manually remove them or
return to ClustalW and generate a new .aln file without line
numbers.
If any extraneous symbols are present following the last sequence,
delete them
- Save
the output file with a .meg extension
C.
Perform clustering analysis in MEGA
- Return
to the initial MEGA window (shown above in III.B.1) and select
"click me to activate a data file".
- Select
the .meg file that you saved in step III.B.5. An "Input
Data" window will appear. Under "Data Type"
select "Protein Sequences" and click OK.
- The window
shown below will appear, with the open
data file indicated at the bottom
- Generate
a phylogenetic tree from the active data file using Phylogeny >
Bootstrap Test of Phylogeny.
At this
point, a number of options can be selected, including UPGMA
Tree, Neighbor-Joining Tree, Minimum Evolution and Maximum
Parsimony. Similarly, in the "Analysis Preferences"
window for UPGMA, Neighbor-Joining, or Minimum Evolution, models
can be selected under Models>Amino Acid. Users are encouraged
to generate trees using a variety of these options. The
general clustering patterns should be similar, regardless
of method.
The
method that best approximates those described in Lindeberg
et al, 2005 (using MEGA 4 rather than MEGA 2.1) involves Neighbor-Joining using the Amino Acid: p-distance model
(the "Analysis
Preferences" window for Neighbor-Joining is shown below)
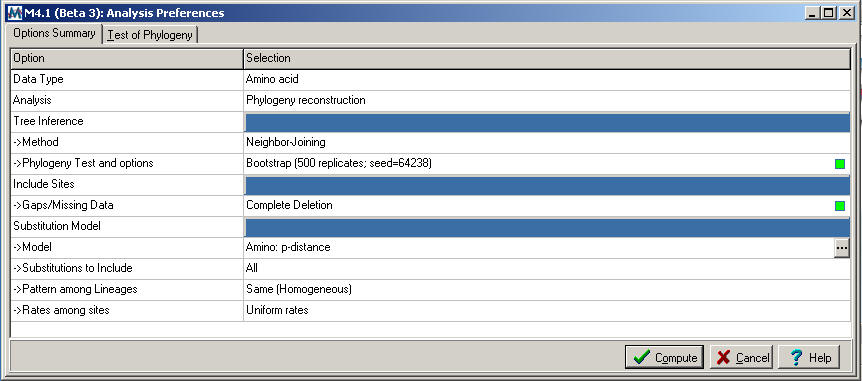
The Bootstrap
consensus tree resulting from Neighbor-Joining analysis for the HopA family is shown below.
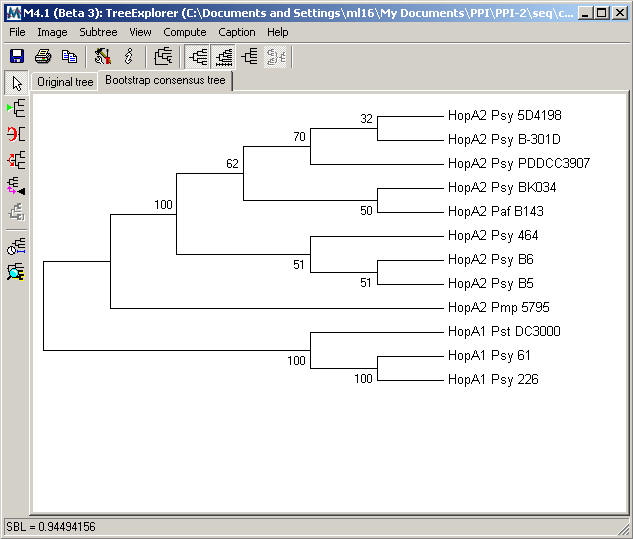
D.
Calculate genetic distance between the new Hop and established
subgroups
The level
of amino acid diversity within and between subgroups was used
as the basis for dividing Hop families (Lindeberg et al. 2005)
As described there, homology families were subdivided if within-group
amino acid diversity was less than 0.75 and between-group amino
acid diversity greater than 0.75, when using a gamma parameter
of 2.25. A cutoff score for MEGA 4 consistent with the previously established subfamily divisions comes to 0.475 when using the Neighbor-Joining Amino acid: p-distance model.
- To calculate
amino acid diversity among the sequences in the active data
file, return to the MEGA window shown in III.C.3. and go to
Distances>Compute Pairwise.
- An "Analysis
Preferences" window will appear similar to that shown
in III.C.4. Select Model>Amino Acid: p-distance if
not already selected.
Click OK
- The resulting
table of pairwise distances for the HopA family is shown below.
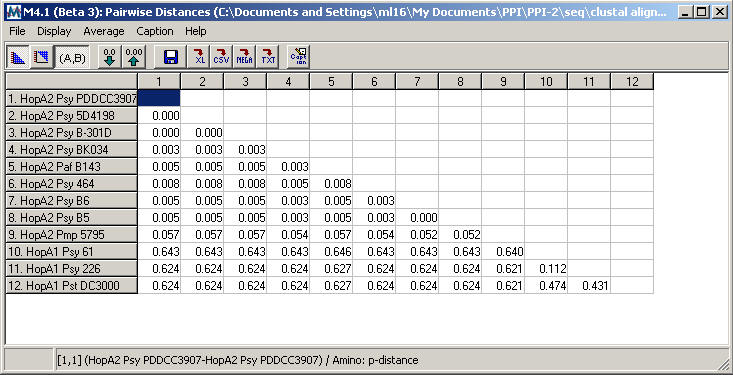
-
Although
the HopA1 subgroup has a higher level of internal diversity
than HopA2, the pairwise distance table shows between-group
diversity > 0.475 and within-group diversity < 0.475,
consistent with the recommendations for subgroup division
described above
|
| |
|